If you want to know about our group’s mission and the work we do, watch this video made by our alumnus, Sam Vaccaro:
Our research has been generously supported by:



Many commercial chemicals pose hazard to human and environmental health. Yet, current legislative programs often do not require that chemicals are tested for safety before they are put into production. In fact, the overwhelming majority of chemicals in use today have never been independently tested for safety. Lax regulations and prohibitive costs of animal testing are the main reasons why chemicals like BPA, phthalates or PBDEs are pulled off the market after prolonged public outcry, only to be replaced with equally toxic substances. Socially and economically, this is not a sustainable approach. To provide a viable alternative to toxicological testing, we develop computer models to study the mechanisms by which toxicants interact with biological targets, such as enzymes or nucleic acids. Our methods focus on design of a priori safer molecules (versus predictions of toxicity after the fact). Leveraging toxicological data, we build algorithms that serve as design vectors for new chemicals, optimizing trade-offs related to safety, depletion and performance.


Our flagship project applies state-of-the-art computational modeling adopted from drug discovery, namely classical and quantum Monte Carlo simulations used conjunction with free energy perturbation calculations, to redesign existing organophosphorus flame retardants and agrochemicals. Our strategy is to optimize their molecular scaffolds so as to minimize toxicity against known biological targets, such as acetylcholinesterase, nucleic acids or androgen and estrogen receptors, while retaining desired function. We can predict functionality using models of thermal and oxidative stability (flame retardants) and by computing biological activity against known targets. Our protocol is designed to yield novel analogs that are safer, functional and can readily deplete from the environment .
Mechanistic investigation of chemical photodegradation to aid in novel pesticide design
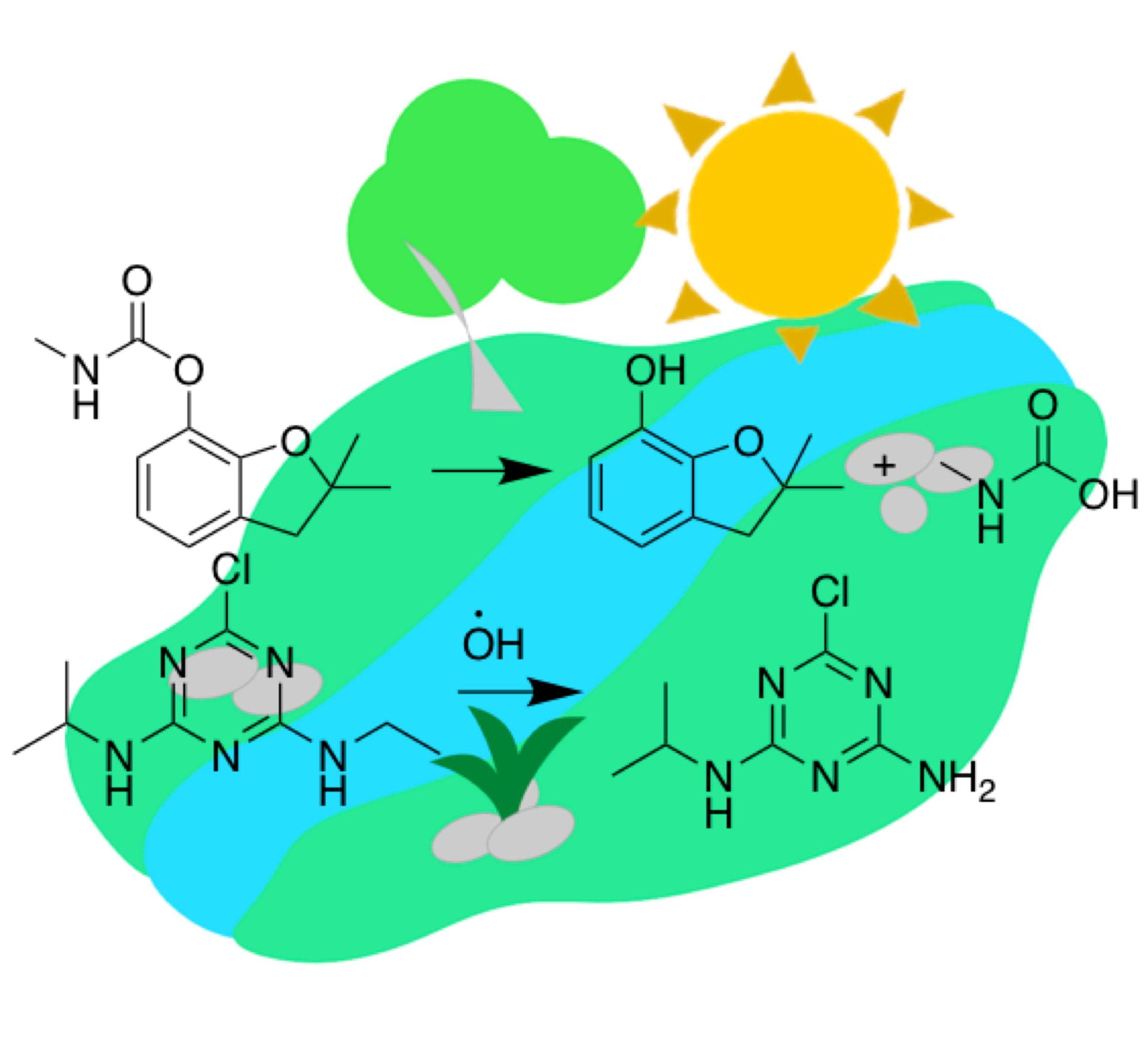 Long-term sustainability goals call for significant changes to the use of chemicals in the environment, especially in the field of crop protection, which is gaining importance as global population grows. In this project, which complements our design efforts for minimal toxicity of pesticides, we are advancing our understanding of pesticide photodegradation mechanisms in troposphere and surface waters. To that end, we have developed a computational framework to study transformations of pesticides with excited triplet states of chromophoric dissolved organic matter, which readily oxidizes substrates based on aminoarene, aryl ether and sulfide cores. This framework balances safety, depletion and performance and has been applied to all relevant pesticides on EPA’s registry. Redox reaction kinetics and thermodynamics are captured using a tandem of DFT reaction-pathway modeling and FMO (frontier molecular orbital) theory. Currently, we are developing an open-access database based on collected data to assist with transformation predictions of existing and with the development of new pesticides. Our strategy for controlled degradability of pesticides is based on methods adopted from drug discovery and state-of-the-art machine learning approaches, exploiting the mechanistic link between pesticide structures and the energetics of their transformations.
Long-term sustainability goals call for significant changes to the use of chemicals in the environment, especially in the field of crop protection, which is gaining importance as global population grows. In this project, which complements our design efforts for minimal toxicity of pesticides, we are advancing our understanding of pesticide photodegradation mechanisms in troposphere and surface waters. To that end, we have developed a computational framework to study transformations of pesticides with excited triplet states of chromophoric dissolved organic matter, which readily oxidizes substrates based on aminoarene, aryl ether and sulfide cores. This framework balances safety, depletion and performance and has been applied to all relevant pesticides on EPA’s registry. Redox reaction kinetics and thermodynamics are captured using a tandem of DFT reaction-pathway modeling and FMO (frontier molecular orbital) theory. Currently, we are developing an open-access database based on collected data to assist with transformation predictions of existing and with the development of new pesticides. Our strategy for controlled degradability of pesticides is based on methods adopted from drug discovery and state-of-the-art machine learning approaches, exploiting the mechanistic link between pesticide structures and the energetics of their transformations.
Upgrading Biomass Using In Silico Designed Ionic Liquids
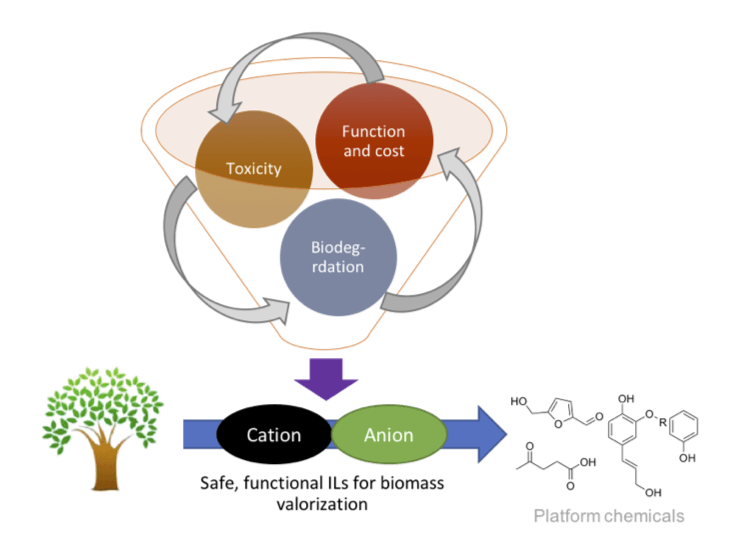 In collaboration with the Voutchkova lab at GWU, we are developing an integrated processes for deconstruction and upgrading of lignocellulosic biomass using ionic liquids designed in silico to be functional and environmentally benign. Despite the significant surge in research aimed at valorization of lignocellulosic biomass, economically-viable and efficient processes are still lacking. Typical workflows for biomass processing require harsh conditions and drastic changes in pH across multiple steps. Ionic liquids have shown theoretical promise for mild pre-treatment of biomass; however, industrial applications are hindered by i) challenges in optimization of numerous physicochemical properties for task-specific ionic liquids, ii) human and environmental toxicity of many ionic liquids, and iii) significantly higher cost compared to conventional solvents. By combining state-of-the-art computational methodology, synthetic and process chemistry, we are iteratively designing novel ionic liquids around existing molecular scaffolds to help overcome these major challenges.
In collaboration with the Voutchkova lab at GWU, we are developing an integrated processes for deconstruction and upgrading of lignocellulosic biomass using ionic liquids designed in silico to be functional and environmentally benign. Despite the significant surge in research aimed at valorization of lignocellulosic biomass, economically-viable and efficient processes are still lacking. Typical workflows for biomass processing require harsh conditions and drastic changes in pH across multiple steps. Ionic liquids have shown theoretical promise for mild pre-treatment of biomass; however, industrial applications are hindered by i) challenges in optimization of numerous physicochemical properties for task-specific ionic liquids, ii) human and environmental toxicity of many ionic liquids, and iii) significantly higher cost compared to conventional solvents. By combining state-of-the-art computational methodology, synthetic and process chemistry, we are iteratively designing novel ionic liquids around existing molecular scaffolds to help overcome these major challenges.
Interdisciplinary Approach to Understand Oxidative Stress in Biological Systems
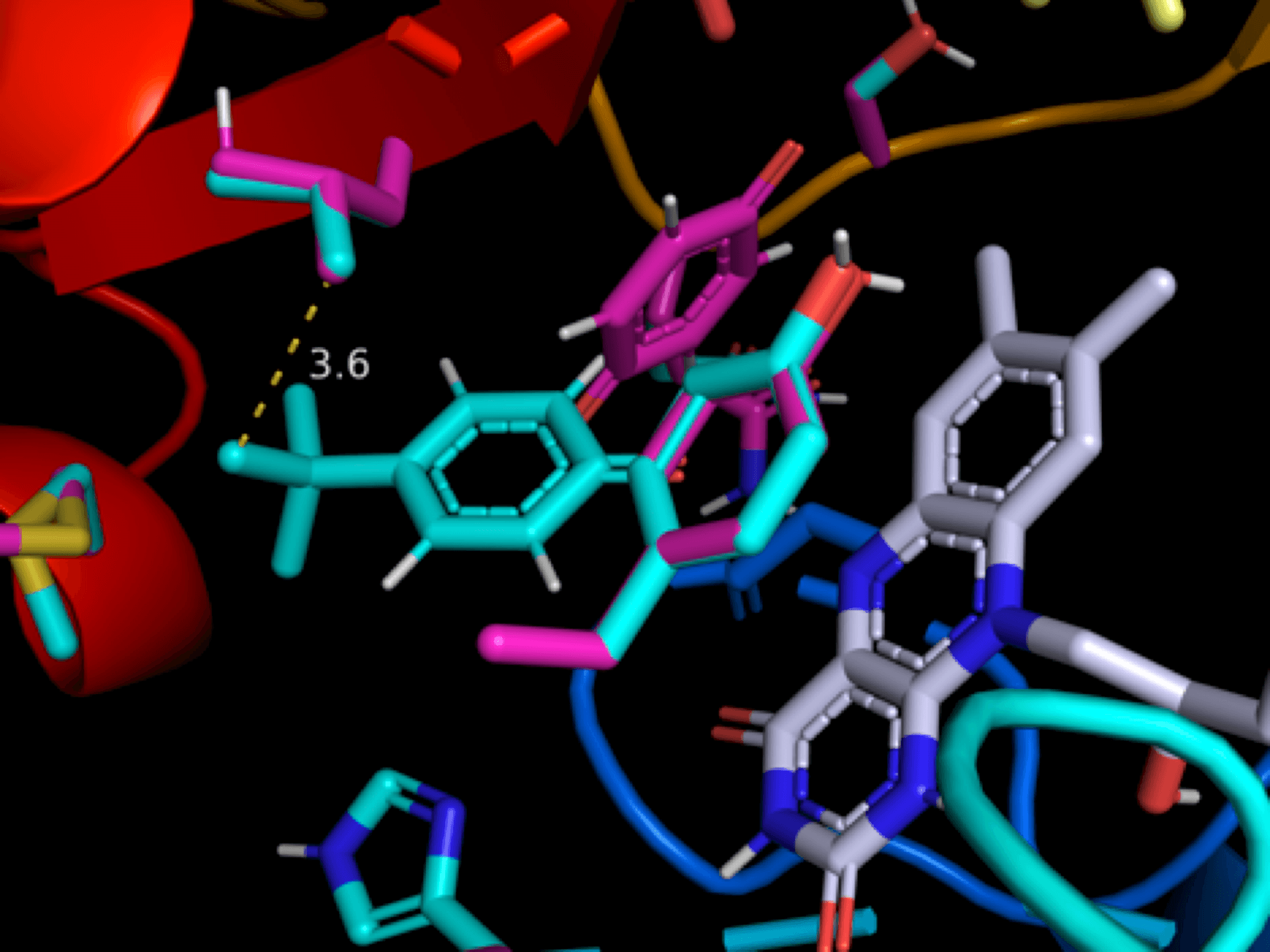 As part of MoDRN, Molecular Design Research Network between Yale U, Baylor U, U of Washington and GWU, we use computer modeling to investigate molecular mechanisms implicated in ROS (reactive oxygen species) generation. Our current focus is on the metabolism of quinone-like agents, which involves enzymatic reduction by one or two electrons to form the corresponding semiquinone or hydroquinone molecules. The consequence of these eductions is the donation of extra electron to oxygen with the formation of a superoxide radical anion. This reduction by a reductase followed by oxidation by molecular oxygen (dioxygen) is known as redox-cycling and continues until the system becomes anaerobic. We use ensemble docking, reaction modeling and FMO (frontier molecular orbital) calculations to support experimental observations.
As part of MoDRN, Molecular Design Research Network between Yale U, Baylor U, U of Washington and GWU, we use computer modeling to investigate molecular mechanisms implicated in ROS (reactive oxygen species) generation. Our current focus is on the metabolism of quinone-like agents, which involves enzymatic reduction by one or two electrons to form the corresponding semiquinone or hydroquinone molecules. The consequence of these eductions is the donation of extra electron to oxygen with the formation of a superoxide radical anion. This reduction by a reductase followed by oxidation by molecular oxygen (dioxygen) is known as redox-cycling and continues until the system becomes anaerobic. We use ensemble docking, reaction modeling and FMO (frontier molecular orbital) calculations to support experimental observations.
Aquatic Toxicity
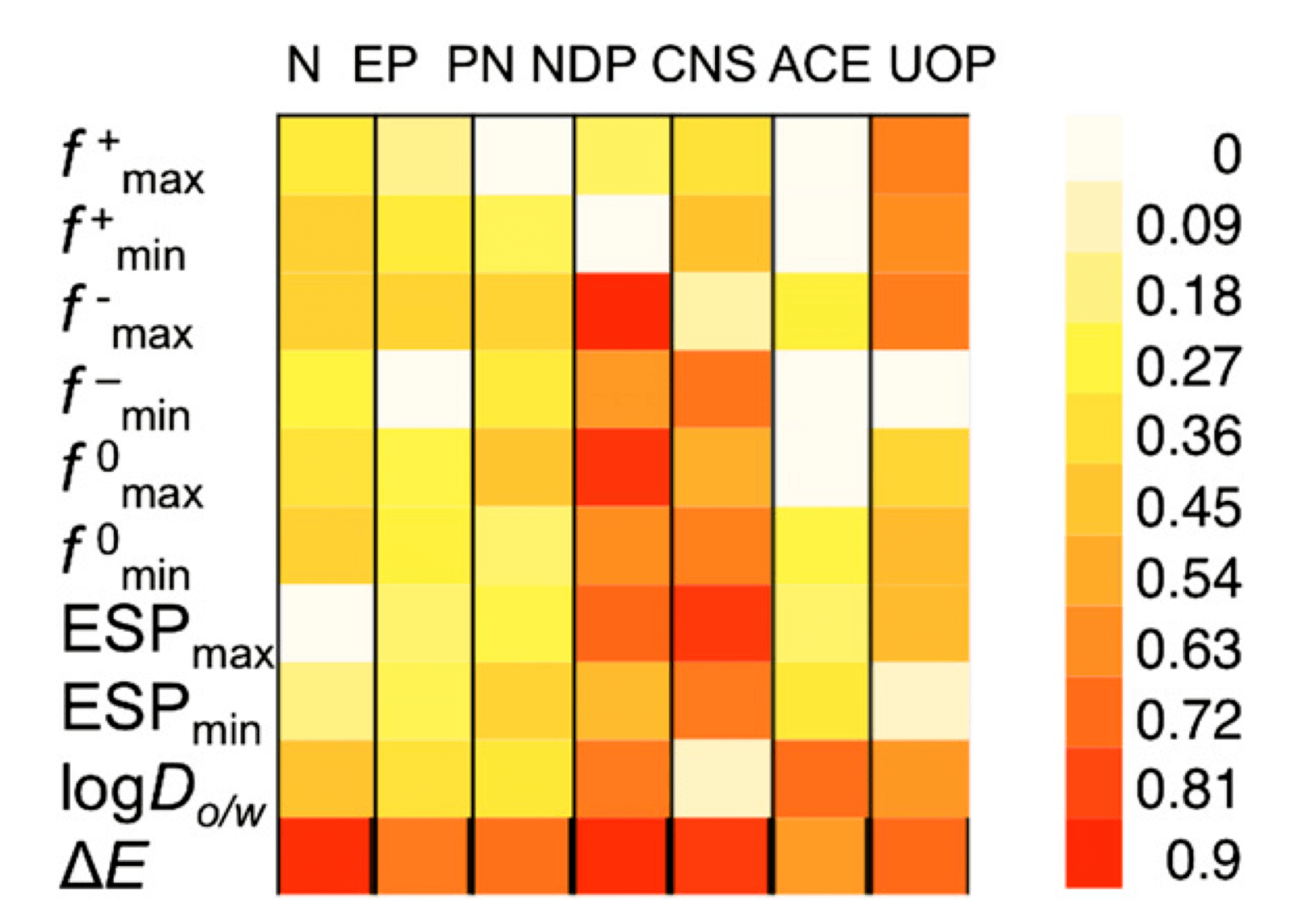 Our ongoing collaboration with the Voutchkova-Kostal Lab at GWU and Brooks Lab at Baylor University aims to improve upon the design guidelines for aquatic toxicity, which we developed in 2012-2015. These guidelines use simple bioavailability and acid-base reactivity metrics, namely the octanol-water distribution coefficient (log D) and the HOMO-LUMO gap, to define a chemical subspace, which corresponds to low probability of toxicity to aquatic species. Our aim is to incorporate metrics for metabolic activation to increase the sensitivity of the model, and extend its predictivity to all categories of concern for aquatic toxicity, as defined by the US EPA.
Our ongoing collaboration with the Voutchkova-Kostal Lab at GWU and Brooks Lab at Baylor University aims to improve upon the design guidelines for aquatic toxicity, which we developed in 2012-2015. These guidelines use simple bioavailability and acid-base reactivity metrics, namely the octanol-water distribution coefficient (log D) and the HOMO-LUMO gap, to define a chemical subspace, which corresponds to low probability of toxicity to aquatic species. Our aim is to incorporate metrics for metabolic activation to increase the sensitivity of the model, and extend its predictivity to all categories of concern for aquatic toxicity, as defined by the US EPA.
Mutagenicity
 Olefins and epoxides are used in paints, coatings, adhesives, textiles, plastics, electronics, etc. They are widely distributed in the environment (over dozen halogenated olefins are in drinking water); yet, many of these chemicals are potent mutagens and carcinogens. Using QM/MM calculations in conjunction with statistical potentials-of-mean force calculations, we can obtain accurate estimates of DNA alkylation rates, which are indicative of mutagenic potential. Combining reactivity parameters with metrics of bioavailability allows us to develop predictive models of mutagenicity. This project serves as a platform for development of novel statistical SMO-SMO (semiempirical molecular orbital) approaches, which are accurate and fast enough to assess all plausible alkylation sites in a nucleic acid target.
Olefins and epoxides are used in paints, coatings, adhesives, textiles, plastics, electronics, etc. They are widely distributed in the environment (over dozen halogenated olefins are in drinking water); yet, many of these chemicals are potent mutagens and carcinogens. Using QM/MM calculations in conjunction with statistical potentials-of-mean force calculations, we can obtain accurate estimates of DNA alkylation rates, which are indicative of mutagenic potential. Combining reactivity parameters with metrics of bioavailability allows us to develop predictive models of mutagenicity. This project serves as a platform for development of novel statistical SMO-SMO (semiempirical molecular orbital) approaches, which are accurate and fast enough to assess all plausible alkylation sites in a nucleic acid target.
Uncertainty in Toxicological Data
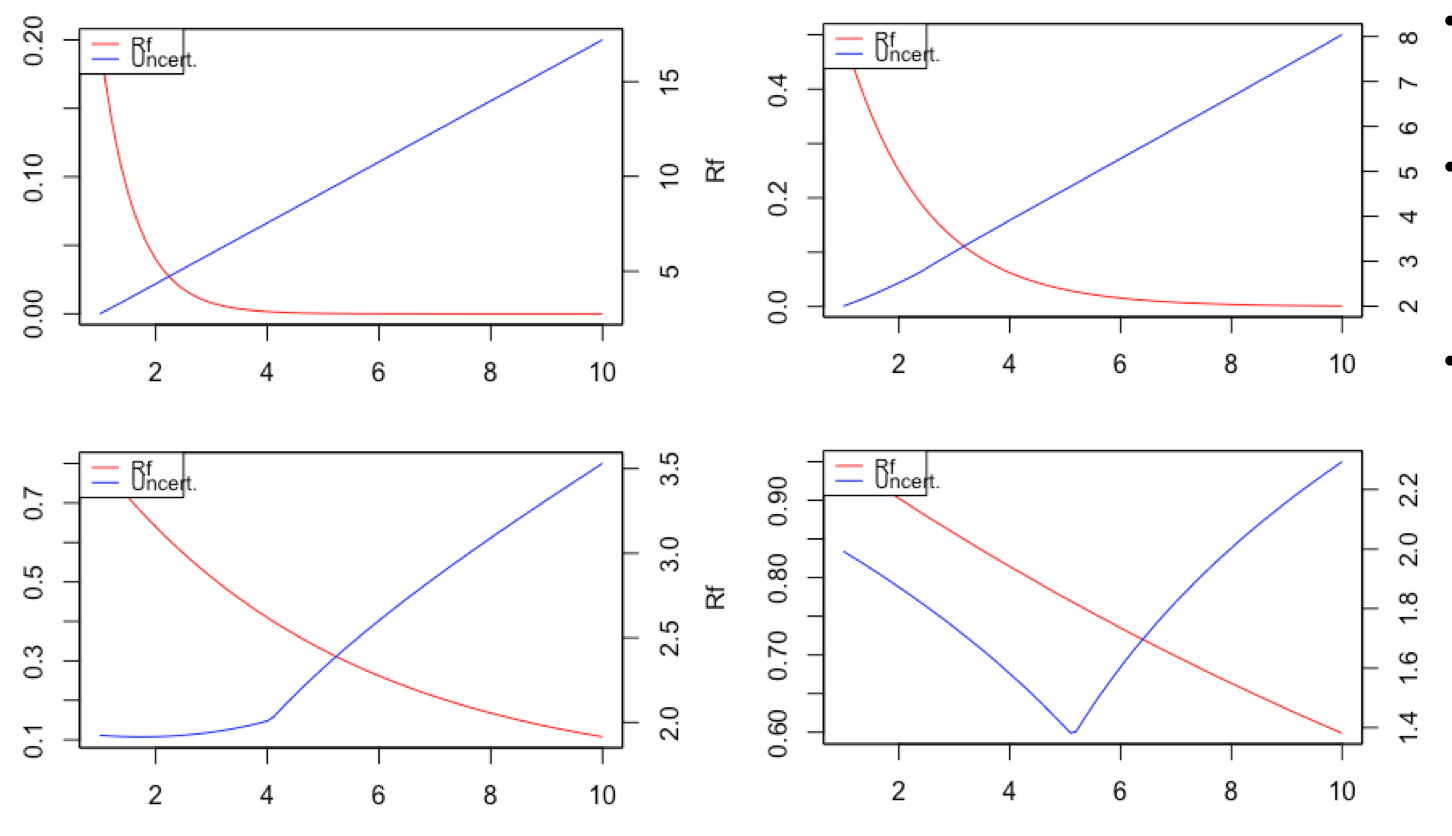 In collaboration with the US EPA, Verisk 3E and Redshift Technologies, we develop and validate novel metrics for assessing uncertainty in toxicological data. This project aims to improve decision making about chemical toxicity when a multitude of conflicting experimental (and predicted) data exists for a given observable. The target users of our approach are model developers and regulators. We are testing several mathematical relationships that not only consider each data point’s intrinsic reliability and relevance but also factor in the overall behavior of the group of equivalent data. Our models are parametrical, capable of reproducing specific (expert) decision making paradigms.
In collaboration with the US EPA, Verisk 3E and Redshift Technologies, we develop and validate novel metrics for assessing uncertainty in toxicological data. This project aims to improve decision making about chemical toxicity when a multitude of conflicting experimental (and predicted) data exists for a given observable. The target users of our approach are model developers and regulators. We are testing several mathematical relationships that not only consider each data point’s intrinsic reliability and relevance but also factor in the overall behavior of the group of equivalent data. Our models are parametrical, capable of reproducing specific (expert) decision making paradigms.